Selected Technologies & Tools
We are answering a broad variety of questions using different technologies and analytical tools. We have described selected technologies and tools here.
Phosphopeptide enrichment
StaGE tips
StaGE (stop-and-go extraction) tips are used for micro solid phase purification (SPE). They can be purchased or made easily in the laboratory. A wide variety of 3M Empore membranes (SAX, SCX, C8 etc.) are available. See how to make a StaGE tip in this video.
Orginal publication on StaGE tips by Yasushi Ishihama can be found following this link: http://www.ncbi.nlm.nih.gov/pubmed/16602707
Stable Isotope Labeling with Amino acids in Cell culture (SILAC)
In this labeling technique one or more amino acids of the nutrient medium is substituted with heavy labeled counterparts. Incorporation of the heavy-labeled amino acids into the proteome is assured by the metabolism of the cells. For HeLa and 293T cells, five passages are sufficient for complete incorporation.
Both Arginine and Lysine are commonly used in SILAC experiments. The reason behind this is the use of trypsin for enzymatic digestion in typical proteomics experiment. Trypsin cleaves the C-terminus of Arginine and Lysine, therefore all generated peptides will be available for quantitation. It is beneficial if the delta mass between light and heavy Arginine and Lysine differs. For example, 13C6-Lysine and 13C615N2-Arginine have mass differences of 6 and 8 Da, respectively, compared to their light-isotope counterparts. Different delta mass for Arginine and Lysine peptides allows the determination of the C-terminal amino acid by measurement of the mass difference for a SILAC peptide pair.
SILAC kits are manufactured by Invitrogen, Pierce and Dundee Cell products.
Suggested SILAC protocols:
http://www.ncbi.nlm.nih.gov/pubmed/18323819
http://www.ncbi.nlm.nih.gov/pubmed/17406521
SILAC websites:
www.silac.org
http://www.biochem.mpg.de/mann/SILAC/index.html
MaxQuant
A Powerful software to search and quantitate LC-MS/MS data. MaxQuant was developed to analyze SILAC experiment from Thermo hybrid FT mass spectrometers but has now gained more features. In addition to ProteomeDiscoverer/MASCOT, the Proteomics Resource Center also uses MaxQUANT with its integrated tandem MS search algorithm, Andromeda.
Link to MaxQUANT/Andromeda report: http://www.ncbi.nlm.nih.gov/pubmed/21254760
Mascot
Mascot is considered the gold standard when it comes to analyzing MS/MS spectra of peptides against protein databases. Mascot and Sequest were some of the first tools that allowed automatic searching of larger sets of MS/MS data. The Proteomics Resource Center is using Mascot v. 2.3, Sequest and Andromeda as database search tools.
ProteomeDiscoverer
This software platform merges many tools for proteomics data analysis, including extraction of MS/MS data prior to search, MS and MS/MS based quantitation, and comparison of multiple analyses. Proteome Discoverer does not perform the database search for aquired MS/MS data, instead it is linked to Mascot that serves this purpose.
Dimethyl labeling-based quantitation
Stable isotope labeling using reductive dimethylation is cost-effective, simple quantitation method able to compete at any level with the other chemical labeling strategies. Simple reagent, formaldehyde, is used to globally label the N-terminus of the peptide and epsilon-amino group of Lysines through reductive amination. This labeling strategy produces peaks differing by 28 Da for each derivatized site relative to its nonderivatized counterpart and 4 Da units for each derivatized isotopic pair. Up to three different experiments can be simultaneously compared using this technique.
The complete experimental procedure for dimethyl labeling can be found here.
Label-free quantitation
Comparison of two or more samples by simply analyzing them once is the most intuitive. However, there are technical obstacles using this strategy. Critical issues are robustness of LC-MS equipment, stability of the mass spectrometer and sample handling. Furthermore, the samples to be compared must be of similar matrix and complexity. If these issues can be controlled it is indeed possible to get reliable results in label-free experiments. The benefit of a label-free strategy is that no labeling is required. Main disadvantages are the difficulty of measuring minor differences or fold changes between samples, as well as the need for at multiple technical LC-MS replicates per sample. Shown below is the distribution of log2 of the ratios of the “protein intensities” for the analysis of the exact same E.coli tryptic digest twice. If the sample is the same and LC-MS analysis is 100% reproducible, it should be expected that all “protein intensity” ratios would be 1 [log2(1)=0]. As it is depicted on the figures below, even though the “protein intensity” has the highest frequency for the value 1, there is a distribution around it as well (A and C). Furthermore, if samples are subjected to some handling prior to analysis, which is most often the case, the distribution will broaden even more. Example is a clean-up using StaGE tips (B and D). Several software solutions are avaiable for label-free quantitation analysis. In the PRC MaxQuant (A and B) and Proteome Discoverer (C and D) are routinely used. While MaxQuant is relying on relative reproducible retention times, Proteome Discover is less sensitive to retention times since ‘abundance’ calculation of a protein is based of the ‘average-intensity-of-the-three-most-intense-peptides’ (see orginal manuscript based on a different hardware concept: http://www.ncbi.nlm.nih.gov/pubmed/16219938).
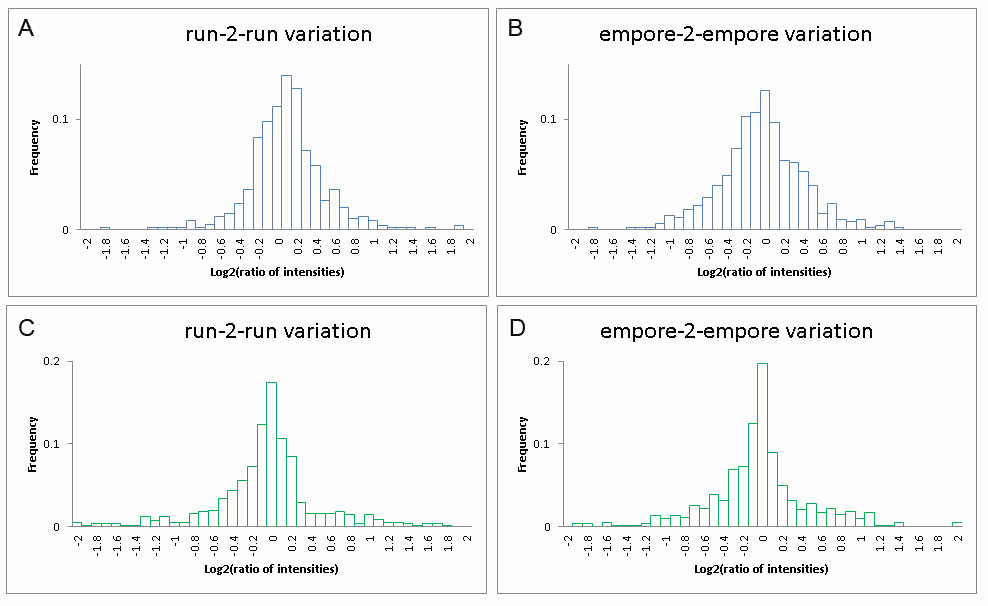 two experiments; A and C are to….. Where as B & C…best case scenario…robustness depends on sample (e.g. salts or detergents) and also amounts loaded
two experiments; A and C are to….. Where as B & C…best case scenario…robustness depends on sample (e.g. salts or detergents) and also amounts loadedAbsolute quantitation (AQUA) of peptides
In short: peptides or peptide fragments of the protein that needs to be quantified are mixed with their stable isotope-labeled (13C15N) counterparts of a known amount. The intensity ratios between these two peptides are measured and the absolute amount of the non-labeled peptide is calculated. The Proteomics Resource Center provides the synthesis of heavy stable isotope-labeled peptides. Please note that utilizing aliphatic amino acids (Leucine, Alanine, and Valine) for introduction of the heavy stable isotope atoms is far less expensive than utilizing amino acids which require side chain reactive group protection (Arginine and Lysine).
Tandem mass spectrometry
Our mass spectrometers are capable of performing different types of MS/MS (tandem MS): collisional induced dissociation (CID), supplemental activation CID, higher energy collisional dissociation (HCD), and electron transfer dissociation (ETD).
iTRAQ/TMT
Isobaric tags for relative and absolute quantitation (iTRAQ) is a quantitation method in which peptide N-terminus and side chain amines are covalently labeled with tags of varying masses. Up to eight different samples or treatments can be analyzed using this technique. After labeling and pooling of all the samples, they are analyzed by LC-MS/MS in a single run. The fragmentation of the attached tags produces a low molecular mass reporter ions that are used to relatively quantify the peptides and the proteins from which they originated. Tandem mass tag (TMT) is a quantitation method in which, as in iTRAQ, peptides are covalently labeled using isobaric tags. The chemical structures of all the tags are identical but each contains isotopes substituted at various positions, so that two out of four regions have different molecular masses in each tag. The combined four regions of the tags have the same total molecular weights and structure, therefore during an LC-MS run peptides labeled with different tags are indistinguishable. Upon fragmentation quantitation data are simultaneously obtained from fragmentation of the tags, giving rise to mass reporter ions.
Sequence databases
More information on databases utilized in Proteomics Resource Center is available here.