Researchers discover how to turn one germ's drug resistance into an Achilles' heel
Key takeaways
- Rifampicin-resistant tuberculosis poses a major global health crisis.
- A new study shows that the most common rifampicin resistance mutation in tuberculosis rewires bacterial gene regulation in a way that creates specific metabolic vulnerabilities.
- The findings could guide the development of combination therapies designed not only to kill drug resistant tuberculosis, but to suppress drug resistance itself.
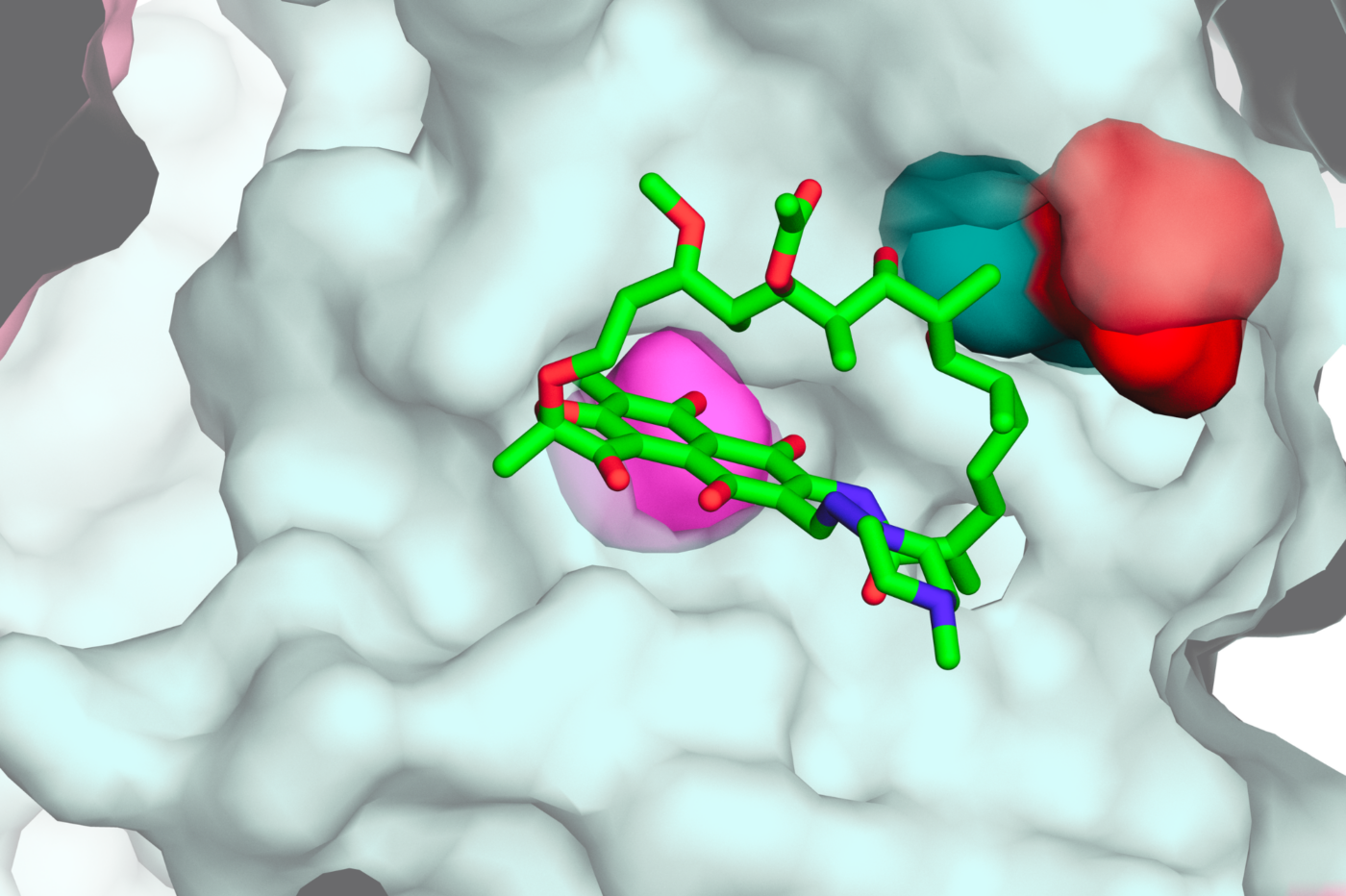
A close-up structural model of a common rifampicin-resistance mutation discovered to create vulnerabilities that could be targeted by future combination therapies. (Credit: Liz Campbell)
Decades of reliance on the antibiotic rifampicin have fueled the rise of drug-resistant Mycobacterium tuberculosis (Mtb). But as the bacterium mutates to protect itself from the drug, it also creates new weak points that other therapies could exploit. Now, a new study in Nature Microbiology shows that the most common rifampicin-resistance mutation slows bacterial RNA polymerase, creating vulnerabilities that future combination therapies may be able to target.
“We’re developing a strategy to stay ahead of drug resistance,” says Jeremy Rock, head of the Laboratory of Host-Pathogen Biology at Rockefeller. “With combination therapies, we could exploit the fact that a mutation that helps the bacteria survive one antibiotic renders it vulnerable to another.”
A global threat
Tuberculosis is the world’s deadliest infectious disease, killing more than one million people each year. Much of modern TB treatment depends on rifampicin, an antibiotic that targets bacterial RNA polymerase, the enzyme that transcribes DNA into RNA and drives gene expression. By binding to the enzyme’s β-subunit, rifampicin prevents the bacterium from properly producing RNA, effectively shutting down its most important functions.
“Rifampicin has historically been part of the backbone of TB treatment,” says Kathryn Eckartt, a PhD Student in the Rock Lab who is now a postdoctoral fellow at Weill Medical College. “So as Rif resistance slowly makes this drug unusable, a lot of lives are in danger.”
For Vanisha Munsamy-Govender, a laboratory manager in the Rock lab who previously worked with TB patients and drug-resistant TB in South Africa, the threat is personal. “I have firsthand insight into the devastating impact these infections have on patients and healthcare systems,” she says. “These experiences shaped my interest in understanding not only how Mtb develops resistance, but also whether resistance mutations create vulnerabilities that could be exploited.”
Previous work had suggested that resistance mutations come with evolutionary trade-offs. When bacteria mutate to evade an antibiotic, this change can disrupt core cellular processes and create weak points. The most common rifampicin-resistance mutation in Mtb, βS450L, illustrates this principle: Elizabeth Campbell and Seth Darst showed it blocks drug binding to RNA polymerase, while Rock’s work with Campbell revealed that the altered enzyme also functions more slowly, frequently pausing, stalling, or prematurely terminating transcription. Bacteria carrying this mutation were also particularly sensitive to disruptions in 150 other genes.
But researchers could not hope to exploit these vulnerabilities until they understood their root cause. If the vulnerabilities were simply a side effect of the sluggish mutant bacteria, they would be difficult to target. But if they were driven directly by the mutant enzyme fundamentally changing how the bacteria transcribes its genetic material, that could provide a specific, mechanistic blueprint for new drugs.
“Basic science gives us the tools to understand what’s changing in these kinds of situations. We hoped that we could then use that knowledge to inform the development of drugs that target weaknesses unique to these mutations,” Eckartt says.
Exploiting new weaknesses
For the study—partly supported by Rockefeller’s SNF Institute for Global Infectious Disease Research, and conducted in collaboration with Shixin Liu‘s Laboratory of Nanoscale Biophysics and Biochemistry—the team compared βS450L, which slows RNAP, against two other common rifampicin-resistance mutations that have the opposite effect and instead produce fast, pause-resistant RNAP. By comparing these distinct mutations, the team showed that the severe metabolic liabilities seen in βS450L are specifically linked to its sluggish transcription machinery—not a mere side effect.
Importantly, even the two fast mutations produced distinct vulnerability profiles from one another, also underscoring that antibiotic resistance is not a one-size-fits-all phenomenon. “Not all rifampicin-resistance mutations behave the same way,” Munsamy-Govender says. “Future therapies may need to account for the specific resistance mutations present in an infection.”
Investigating further, the team found that bacteria carrying βS450L become unusually dependent on pathways involved in producing thiamine and branched-chain amino acids. They ultimately traced this dependency to the mutant’s pause-prone RNAP, which disrupted a regulatory RNA sequence positioned before the ilvB1 gene. Under normal conditions, this sequence acts like a molecular switch that senses nutrient levels and determines whether transcription continues so that bacteria can produce more IlvB1, an enzyme needed for creating new amino acids under stress. In βS450L bacteria, RNAP would often stall or shut down at this regulatory checkpoint, preventing Mtb from activating the pathway when nutrients were scarce. The researchers suggest the defect represents a broader regulatory collapse.
The researchers predicted that Mtb with the βS450L mutation would therefore be particularly vulnerable to disruptions in the ilvB1 pathway. To test this, they exposed the bacteria to chlorflavonin, a compound that specifically targets the IlvB1 enzyme. As expected, the βS450L mutant proved far more sensitive to the drug than the faster rifampicin-resistant strains. The finding is especially striking because IlvB1 is already considered a promising TB drug target.
“We showed that the most common rifampicin-resistance mutation does more than just help the bacteria evade antibiotics—it creates new weaknesses,” Munsamy-Govender says. “Exploring these trade-offs could help guide the development of combination therapies designed not only to treat TB, but also to limit the emergence and persistence of drug resistance itself.”
While the work points toward a promising new strategy for confronting multidrug-resistant TB, the researchers caution that it remains at an early stage. The chlorflavonin compound used in the study is not yet suitable as a treatment, and substantial additional work will be needed to determine whether these vulnerabilities can be safely and effectively exploited in patients.
“But our work shows that if we understand the biology deeply enough, drug discovery can build on that understanding to design rational combination therapies for TB,” says Rock.


Key takeaways
- Rifampicin-resistant tuberculosis poses a major global health crisis.
- A new study shows that the most common rifampicin resistance mutation in tuberculosis rewires bacterial gene regulation in a way that creates specific metabolic vulnerabilities.
- The findings could guide the development of combination therapies designed not only to kill drug resistant tuberculosis, but to suppress drug resistance itself.