Evidence builds for the role malfunctioning protein removal systems play in neurodegenerative diseases
Key takeaways
- A new study identifies genetic mutations that interfere with the body’s ability to degrade the protein clumps characteristic of neurodegenerative diseases like Parkinson’s and Alzheimer’s.
- These mutations cause the PI31 protein to malfunction, rendering it unable to transport protein-degrading machines called proteasomes to synapses, which results in protein clumps.
- The study provides direct evidence for the clinical relevance of the PI31 proteasome transport mechanism.
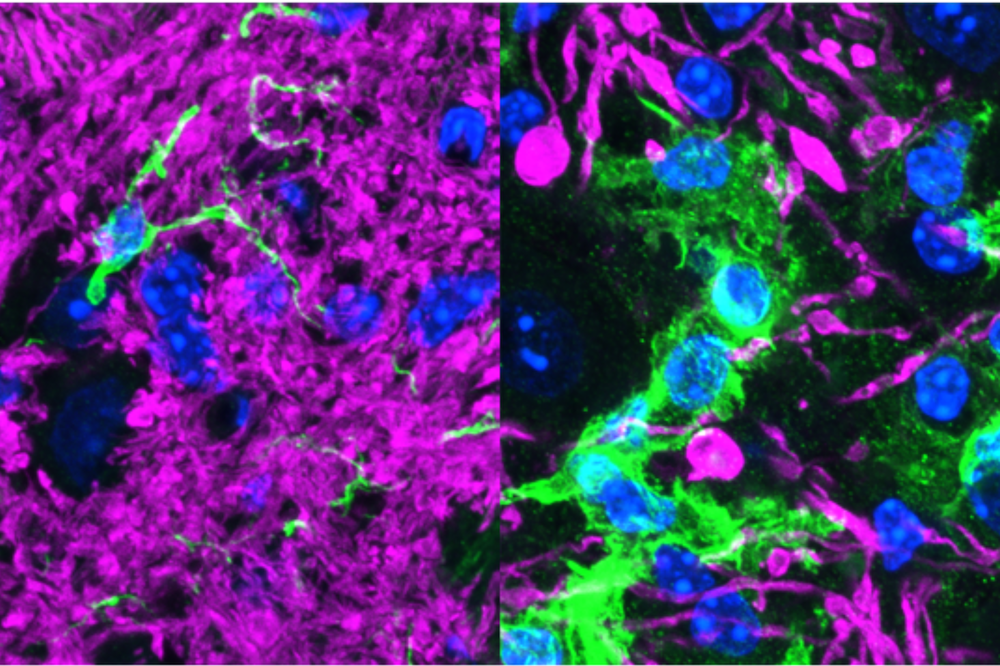
Left: Nerve fibers from healthy brain tissue are shown in magenta with support cells called glia in green. Right: Neurons deprived of PI31 are swollen and damaged. Glia are activated and enlarged as they try to remove faulty connections between cells. (Credit: Steller lab)
Though protein clumps associated with Alzheimer’s and Parkinson’s were discovered over a century ago, we remain largely unable to prevent them from forming or eliminate them from the brain. And though a variety of therapies have taken aim at tau tangles, beta-amyloid plaques, and Lewy bodies, among other notorious aggregates, none have been very effective at stopping disease progression.
Rockefeller’s Hermann Steller and his team in the Strang Laboratory of Apoptosis and Cancer Biology have long been focused on understanding how the cell’s protein-degrading machines, called proteasomes, are regulated. His lab discovered that a transporter protein termed PI31 shuttles proteasomes over long distances between the nerve cell body to synapses. When this system fails, synapses become depleted of degradative capacity, and proteins that should have been eliminated accumulate. As a result, synaptic communication breaks down, protein clumps form, and neuronal health deteriorates.
Now a new study in Nature Communications, led by researchers from University College London and contributed to by Steller’s lab, has identified mutations in PSMF1, the gene that produces PI31, which cause the protein to malfunction. Moreover, the scientists demonstrated that these mutations cause a spectrum of severe, very early onset neurological disorders.
“Based on work in model organisms, we had originally proposed that insufficient proteasome transport causes a local shortage of proteasomes at synapses, and that this may be the root cause of age-related neuronal degeneration,” says Steller. “The new study is an exceptionally important and exciting advance, as it provides direct evidence for the clinical relevance of the PI31 proteasome transport mechanism.”
The role of transport dysfunction
Steller’s work on PI31 dates back some 15 years, when his lab discovered that PI31 boosts the activity of proteasomes and handles their transport logistics, thereby governing their distribution in different nerve cell compartments.
Nerve cells are highly polarized and have interconnected fibers that can extend long distances from the cell body. For brain cells to properly communicate with each other, proteins are continuously manufactured and degraded at synapses. In 2019, Steller’s lab found that knocking out PSMF1, the gene that produces PI31, led to synaptic dysfunction and neuronal degeneration. And in 2025, the researchers discovered that in animal models, boosting PI31 can prevent neuronal degeneration and restore synaptic function.
“Collectively, our work suggests that the progressive failure of synapses in aging and diseased brains is the result of impaired local degradation of proteins that are critical for synaptic function,” says Steller.
A new therapeutic approach
In the current study, UCL first author Francesca Magrinelli and her colleagues examined the genetics of 25 individuals from 18 families of diverse ethnic backgrounds, finding that PSMF1 mutations were associated with a range of severe, and even lethal, neurological disorders that spanned ages from infancy through adulthood.
As part of this study, Jose Rodriguez, a postdoctoral associate in the Steller lab, probed the pathological consequences of these mutations in animal models. He found that PI31 deficiency caused movement and behavioral problems, followed by neurodegeneration.
“Francesca’s study is an important step toward developing new therapeutic strategies to preserve cognitive function in the aging and diseased brain,” Steller says. “In my view, the protein aggregates that define neurodegenerative diseases are the consequence, and not the primary cause of these diseases. Therefore, increasing the activity of proteasomes at synapses is expected to assure the removal of all unwanted proteins, and plaques will never form.”

Key takeaways
- A new study identifies genetic mutations that interfere with the body’s ability to degrade the protein clumps characteristic of neurodegenerative diseases like Parkinson’s and Alzheimer’s.
- These mutations cause the PI31 protein to malfunction, rendering it unable to transport protein-degrading machines called proteasomes to synapses, which results in protein clumps.
- The study provides direct evidence for the clinical relevance of the PI31 proteasome transport mechanism.


