Unique mutation reveals a new role for well-known DNA-repair gene
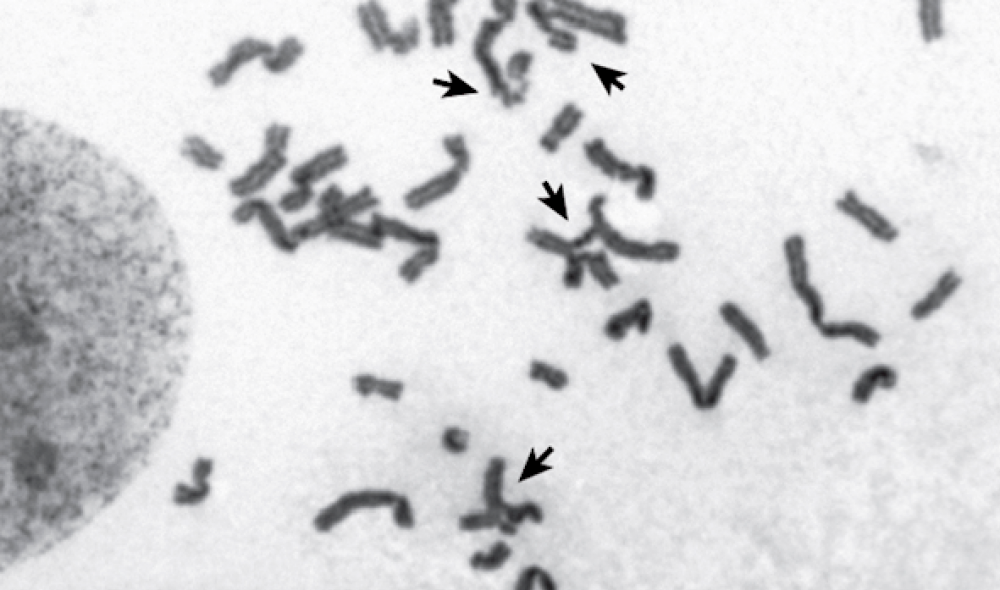
Without BRCA2, a cell cannot repair its damaged chromosomes (shown by arrows).
As far as genes go, BRCA2 is quite famous. Its notoriety comes not from its function—BRCA2 is critical for fixing damaged DNA—but rather from what happens when that function is lost. Mutations in BRCA2 have been linked to breast and ovarian cancer as well as to Fanconi anemia, a rare bone-marrow and cancer predisposition disease in children.
Studying two sisters with unique BRCA2 mutations, researchers have now found that the gene carries out a previously undescribed task to ensure a smooth DNA-repair process. These findings, published recently in Genes and Development, expand scientists’ understanding of BRCA2-related disease as well as the basic processes that keep our DNA safe.
Two sisters, one mutation
Agata Smogorzewska, head of the Laboratory of Genome Maintenance, studies Fanconi anemia (FA) as part of her research into the mechanisms that maintain cellular health. “A lot of our work starts with patients,” says Smogorzewska; and her latest investigation was no exception.
It began when Kim Rickman, a biomedical fellow in her lab, identified a pair of sisters with FA-like developmental problems that lacked a genetic explanation. Despite her extensive research on BRCA2, Smogorzewska did not initially consider the gene a likely culprit. Most children with FA due to BRCA2 mutations develop fatal cancers by the age of five; these women had reached their twenties and were cancer free.
“We sequenced the DNA of both sisters and my analysis didn’t reveal anything because I wasn’t looking at BRCA2,” says Smogorzewska. “Fortunately, Kim was more studious.”
FA has been linked to mutations affecting both copies of the BRCA2 gene, whereas adult cancers typically arise when only one copy of the gene is compromised. Rickman’s analysis showed that the sisters did, indeed, have mutations on both copies of BRCA2. And while this finding clarified the source of the sisters’ condition, it also presented a new set of questions.
Why didn’t these women develop tumors? What made them different from other individuals with two BRCA2 mutations? To find the answers, Smogorzewska and Rickman recreated the sisters’ mutation in individual cells using CRISPR/Cas9 technology and analyzed its effects on a microscopic scale.
A multipronged DNA protector
Genetic mutations come in myriad forms. Depending which bits of DNA are compromised, different components of cellular function are lost. One common BRCA2 mutation, for example, interferes with cells’ ability to repair double-stranded damage to DNA. Under normal circumstances, cells can patch up DNA breaks by copying the similar DNA from a different strand—a process called homologous recombination. Historically, the harmful effects of BRCA2 disorders have been ascribed to the breakdown of this process.
When Rickman studied cells with the sisters’ mutation, however, she found that homologous recombination was largely intact. Yet, not everything functioned smoothly. Another facet of genetic upkeep is to correct lesions on DNA that prevent its strands from separating. Normally, cells can fix these lesions, known as interstrand crosslinks; but in the sisters’ cells, the reparative process proceeded poorly.
Since the cells could perform homologous recombination sufficiently, but not interstrand crosslink repair, the researchers concluded that BRCA2 plays two distinct roles in mending DNA. And, Rickman found, those different roles are performed by different regions of the protein. “BRCA2 is a very large protein and it’s difficult to dissect what the different regions are doing exactly,” says Smogorzewska. “But here we could show that a particular piece of protein that has been previously shown to bind to DNA is especially important for a particular function.” This new insight about BRCA2 functions suggests that defects in BRCA2 that largely spare homologous recombination protect against early cancers. However, due to the disruption in interstrand crosslink repair, they still result in Fanconi anemia.



