First structural map of the cystic fibrosis protein sheds light on how mutations cause disease
Rockefeller scientists have created the first three-dimensional map of the protein responsible for cystic fibrosis, an inherited disease for which there is no cure. This achievement, described December 1 in Cell, offers the kinds of insights essential to better understanding and treating this often-fatal disease, which clogs the lungs with sticky mucus, leading to breathing problems or respiratory infections.
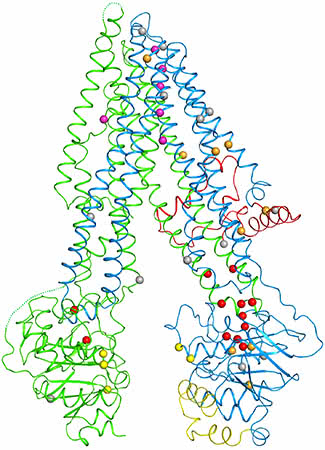
A weak point: When the researchers plotted the locations of mutations (spheres) within the cystic fibrosis protein, they found that many are clustered at a vulnerable joint (lower right).
“With the three-dimensional structure, which we have resolved down to the level of atoms, we can say more about how the cystic fibrosis protein works normally and visualize how it becomes altered in patients,” says senior author Jue Chen, William E. Ford Professor and head of the Laboratory of Membrane Biology and Biophysics.
Chen and first author Zhe Zhang, a postdoc in her lab, plotted out the locations of disease-causing mutations within the structure and revealed a vulnerable spot in the protein that appears responsible for many cases of the disease. “By examining what goes wrong here and at other sites, we hope it may become possible to devise treatments that correct these errors,” Zhang says.
Molecules on ice
The protein in question, the cystic fibrosis transmembrane conductance regulator (CFTR), forms a channel on the surfaces of cells that allows particles of chloride, a component of salt, to pass through the cell membrane. Because the distribution of salt affects the movement of water, a disruption to the channel dehydrates the mucus lining certain organs—including the lungs, where the accumulation of thick mucus can allow bacteria to flourish and potentially lead to life threatening complications.
So far, scientists have identified hundreds of mutations in CFTR’s gene that can cause this disease. Thus far, it hasn’t been entirely clear how these errors interfere with the channel since researchers have been unable to determine its structure.
The main problem was that CFTR resisted scientists’ efforts to turn it into crystals—an essential step in the standard method for determining the structure of a molecule. So instead, Chen and Zhang used an advanced technique known as cryo-electron microscopy, in which molecules are frozen in ice without needing to be made into crystals.
With state-of-the-art equipment installed at the Evelyn Gruss Lipper Cryo-Electron Microscopy Resource Center in 2014, Chen and Zhang took pictures of nearly a million CFTR molecules frozen in a thin layer of ice. By compiling the two-dimensional snapshots, they created the first complete three-dimensional structure of the cystic fibrosis protein.
Opening the door
Within the structure, which resembles a clothespin embedded within the cell’s protective membrane, the researchers saw a glimpse of the mechanics by which the channel opens and closes. These included the first glimpse of a mysterious chunk of CFTR, known as the R domain. It sits between the prongs of the clothespin, keeping the channel closed by preventing the normal movement of the prongs.
“Seeing the first glimpse of the whole molecule felt like a privilege,” says David Gadsby, Patrick A. Gerschel Family Professor at Rockefeller and head of the Laboratory of Cardiac and Membrane Physiology. Gadsby, who has been studying the CFTR protein for 25 years, was not directly involved in determining the structure but helped Chen and Zhang interpret their findings. “It was fantastic,” he says. “It was as if Mother Nature had finally opened a door that had long been shut.”
Mapping mutations
Within the new structure, Zhang and Chen pinpointed 46 sites that are altered by disease-causing mutations. A striking trend emerged: One half of the channel bore many more of these errors than the other, including one responsible for 70 percent of disease cases. An inspection of the map revealed that this part of the protein contains a weak joint more vulnerable to disruption.
“We think this is a good place to start looking for ways to treat cystic fibrosis at its source,” says Chen, who is also a Howard Hughes Medical Institute investigator. “If I were a biochemist developing drugs, I would look for something that could act a bit like glue to strengthen this weak point within the protein.”
 Cell 167, 1586–1597 Cell 167, 1586–1597Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator Zhe Zhang and Jue Chen |