Researchers find combined effects of two genes responsible for premature skull fusion in infants
During the first year of life, the human brain doubles in size, and continues expanding through adolescence. The loosely connected bony plates of the young skull accommodate this growth. But sometimes, these bones fuse too early, a disorder known as craniosynostosis. This disorder can produce facial and skull deformities, and, in some cases, put potentially damaging constraints on a young brain.
A research team led by Rockefeller University President Richard P. Lifton has identified mutations responsible for a type of craniosynostosis that affects the joining of bones along the top of an infant’s skull. The results were published September 8 in eLife.
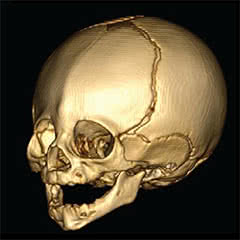
Premature fusion: In one type of midline craniosynostosis, the two plates at the front of an infant’s head fuse before they should, forming a boney ridge down the forehead. In addition to skull abnormalities, this condition can lead to neurodevelopmental problems. (Image credit: Andrew Timberlake)
“This discovery will lead to new ways to diagnose this disorder, as well as help to counsel couples at risk of passing it on to their children. It also has much broader relevance for understanding the genetics of complex traits, including many human diseases,” says Lifton, who is head of Rockefeller’s Laboratory of Human Genetics and Genomics and was a Howard Hughes Medical Institute investigator prior to moving to Rockefeller. The research took place at Yale University, where Lifton was formerly Sterling Professor of Genetics.
Lifton and his team at Yale, including John Persing and colleagues in the Section of Plastic and Reconstructive Surgery, found that midline craniosynostosis arises in people who carry rare mutations in one gene along with common mutations near a second. As such, this disorder is a unique example of how an interaction between genes can contribute to disease.
Sporadic inheritance
Midline craniosynostosis occurs when the bones in front of or behind the soft spot atop a baby’s skull fuse early, producing a ridge or other distortions and, in some cases, neurological problems. First author Andrew Timberlake, an M.D.-Ph.D. student in Lifton’s lab at Yale, used social media to recruit many of the 191 families who participated in this study.
By plotting out the inheritance of the disorder through the families, the team noticed that it showed up unpredictably within them. Clearly, they thought, something more than simple dominant or recessive inheritance was at play. When they sequenced the participants’ protein-coding genes, or exomes, mutations in one gene, SMAD6, caught their attention. This made sense; SMAD6 is a protein that inhibits so-called BMP signaling, which promotes bone formation. But not everyone carrying these rare SMAD6 mutations had midline craniosynostosis. In fact, none of the parents who shared the SMAD6 mutation with their affected children had a history of craniosynostosis—a finding that initially surprised researchers.
A partner in crime
The team then looked to find mutations that might further encourage bone formation. Some previous work implicated common changes near one such gene, BMP2, so they looked for these variations among the families.
“It was amazing to then find that the affected children had inherited both the rare SMAD6 mutation and the common BMP2 variant. In each case, the SMAD6 mutation came from one parent and the BMP2 variant came from the other parent, explaining why neither parent had craniosynostosis,” Timberlake says.
The researchers believe that the risk variants near BMP2 increase the levels of the bone-promoting BMP signaling, thereby amplifying the loss of SMAD6’s ability to put on the brakes. The result: The gap between the skull bones fuses ahead of schedule.
Among the families studied, those who carried the rare, damaging SMAD6 mutation plus a common BMP2 variant always had midline craniosynostosis, while those with only a SMAD6 mutation, but no BMP2 risk allele, were much less likely to suffer from the disorder.
New insight on disease risk
The researchers suggest that a similar dynamic may be at play in other rare genetic disorders that don’t appear to follow classical Mendelian inheritance patterns.
“Our results offer a clear demonstration of the interaction between rare and common variants,” Lifton says, “offering one explanation to a lingering question in genetics: Why do some individuals with potent rare mutations develop disease, while others with the same mutations do not?”
Patients for the study were recruited from the Yale Section of Plastic and Reconstructive Surgery, the Cranio Kids-Craniosynostosis Support Group, and the Craniosynostosis-Positional Plagiocephaly Support Group. This study was supported by the Yale Center for Mendelian Genomics and the Maxillofacial Surgeons Foundation/ASMS, which are supported by the NIH, as well as the NIH Medical Scientist Training Program.
 eLife 2016;10.7554/eLife.20125 eLife 2016;10.7554/eLife.20125Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles Andrew T. Timberlake, Jungmin Choi, Samir Zaidi, Qiongshi Lu, Carol Nelson-Williams, Eric D. Brooks, Kaya Bilguvar1, Irina Tikhonova, Shrikant Mane, Jenny F. Yang, Rajendra Sawh-Martinez, Sarah Persing, Elizabeth G. Zellner, Erin Loring, Carolyn Chuang, Amy Galm, Peter W. Hashim, Derek M. Steinbacher, Michael L. DiLuna, Charles C. Duncan, Kevin A. Pelphrey, Hongyu Zhao, John A. Persing, and Richard P. Lifton |


