Mechanism of mutant histone protein in childhood brain cancer revealed
Most cancer treatments are blunt. In an attempt to eradicate tumors, oncologists often turn to radiation or chemotherapy, which can damage healthy tissue along with the cancerous growths. New research from C. David Allis’s laboratory at Rockefeller University may bring scientists closer to designing cancer therapeutics that can target tumors with pinpoint accuracy.
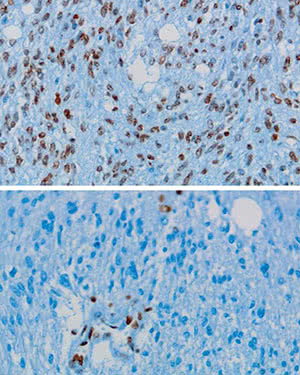
Histone effect. In a rare form of pediatric brain cancer called DIPG, a mutated histone H3 protein inhibits an enzyme that normally keeps cell growth in check. Rockefeller scientists found a global reduction in the levels of methylation (dark areas) of normal H3 histones when small amounts of the mutant H3 were added to normal human cells (bottom).
Their findings, published online today in Science Express, follow a recent series of discoveries by several international genome sequencing consortiums that directly links a mutated histone protein to a rare brain stem cancer in children called DIPG. Collectively, these studies represented the first time scientists had linked a histone mutation to a disease, and piqued the interest of Peter Lewis, a research associate in Allis’s Laboratory of Chromatin Biology and Epigenetics, who spearheaded these new studies.
Together with DNA, histones comprise the gene packaging material called chromatin. The mutation occurs on histone H3, and involves the remarkably specific substitution of one amino acid, lysine, for another, methionine, at a key position on the histone’s tail, “silencing” the associated gene. Normally, gene silencing arises when an enzyme called a methyltransferase, containing a structural region called the SET domain, attaches a methyl chemical group to the lysine at position 27 in the H3 tail. This highly specific chemical reaction, called methylation, is disrupted by the replacement of the lysine with methionine, which could result in gene mis-regulation.
Lewis and his colleagues looked at human DIPG tumors that contained the lysine-to-methionine substitution and determined that mutated histone H3 comprised anywhere from 3.6 percent to 17.6 percent of total H3 in DIPG samples. They also found a global reduction in the levels of methylation of normal H3 histones when small amounts of the mutant H3 were added to normal human cells.
“I have often said, ‘Every amino acid in histones matters,’” says Allis, who is the Joy and Jack Fishman Professor. “These studies underscore just how true that may be.”
The researchers went on to demonstrate that the reduction in methylation of normal H3 histones results from interference with activity of a methyltransferase called PRC2 by the mutant histone. Methylation of normal H3 by PRC2 leads to repression of genes involved in cellular growth pathways. Without methylation, genes involved in these pathways likely become activated, promoting the growth of tumors in DIPG. Allis and Lewis received funding from the National Institutes of Health and the Starr Cancer Consortium. Key collaborators in this work included Oren Becher at Duke University Medical Center and Tom Muir at Princeton University and their colleagues.
“Our findings provide us with a useful tool for probing biological processes,” says Lewis. “This also tells us how to inhibit enzymes, which could lead to the development of pharmaceuticals that mimic the action of these mutants.”
“We now have a model for the promotion of brain stem cancers through aberrant epigenetic silencing through the inhibition of PRC2 by a mutant histone,” says Allis. “We have uncovered a potentially useful mechanism to exclusively inhibit individual SET-domain methyltransferases, and conceivably other chromatin-modifying enzymes, implicated in a variety of malignancies.”
 |
Science online: March 28, 2013 Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma Peter W. Lewis, Manuel M. Müller, Matthew S. Koletsky, Francisco Cordero, Shu Lin, Laura A. Banaszynski, Benjamin A. Garcia, Tom W. Muir, Oren J. Becher and C. David Allis |


