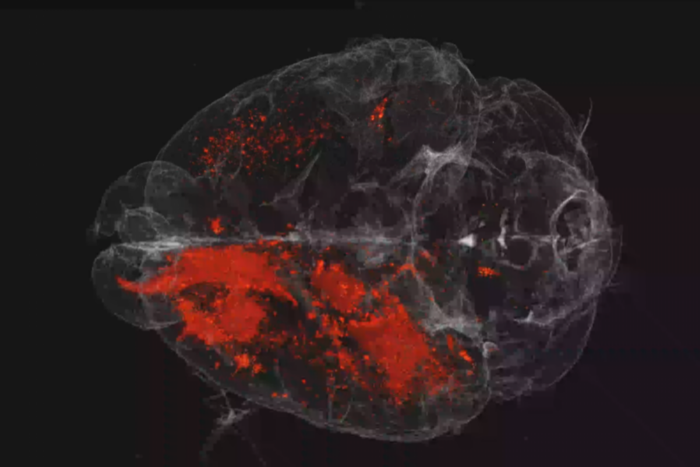
A pioneering way to target the culprit behind a deadly liver cancer
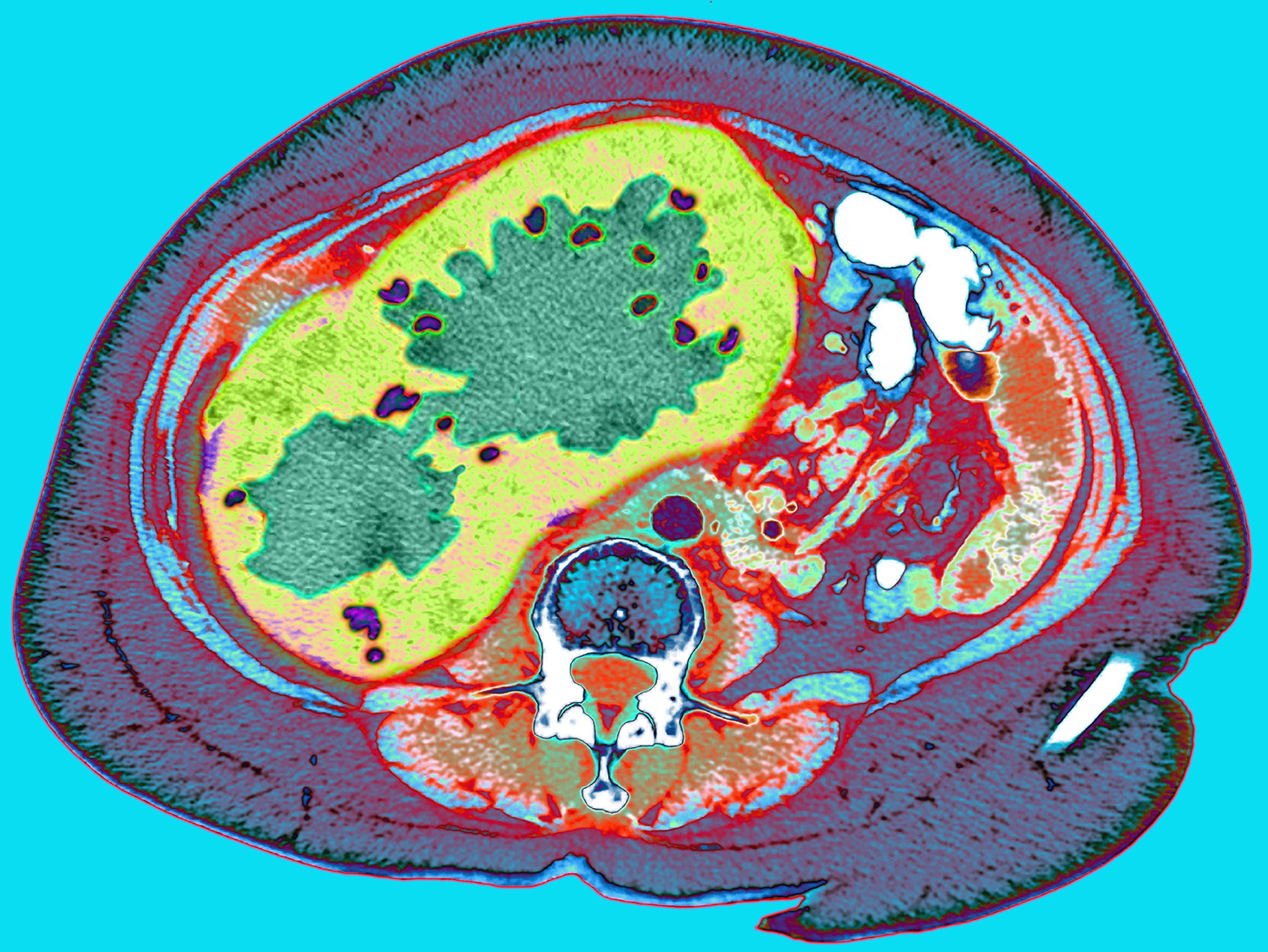
CT scan through a cancerous liver (Du Cane Medical Imaging LTD / Science Photo Library)
Cell division is the generative spark of nearly every lifeform on Earth. But if healthy growth goes unchecked, cell division can turn lethal, overwhelming the organism. Such is the case with so-called oncogenes. When altered by a mutation, these growth-moderating genes go haywire, producing a geyser of cancer cells as a result. Oncogenes are also insidiously adept at generating tumors that over time become genetically “independent” from their origin, so it has been exceedingly difficult for scientists to shut down their replication at the source.
Now Rockefeller University researchers have discovered a new way to target the oncogene behind a rare and often deadly liver disease using small interfering RNAs (siRNAs), an innate mechanism for silencing gene expression. As described in Molecular Therapy, the researchers slipped siRNAs inside fibrolamellar hepatocellular carcinoma (FLC) cells through a surface receptor, where they blocked the oncogene from producing illness-inducing proteins that lead to tumor formation.
It’s the first time that siRNAs have been used to hobble the progression of FLC, says first author Christoph Neumayer, a Ph.D. student in Rockefeller’s Laboratory of Cellular Biophysics, headed by Sanford M. Simon.
But the findings have wider applications as well. “That’s the really exciting part,” says Neumayer. “This is proof of concept that siRNAs can be used for FLC, as well as adult liver cancers, which are much more common, and other tumor types elsewhere in the body.”
The fusion oncogene
FLC is caused by the fusion of two genes located on chromosome 19: DNAJB1, which produces heat-shock proteins that encourage cell homeostasis, and PRKACA, which produces an enzyme called kinase A that’s key to cellular metabolic function. The resulting fusion, known as DNAJB1::PRKACA, brings together the proteins and a catalytic subunit of the kinase. This dysfunctional union promotes FLC formation, but how, exactly, is still unknown.
Researchers in Simon’s lab discovered that this fusion caused FLC in 2014, several years after Simon’s own teenage daughter, Elana, was diagnosed with liver disease. After having the resulting tumor removed—the only treatment option available to people with FLC—Elana went on to collaborate with her father to unearth its cause, publishing the results in Science.
Since then, Simon and his group have continued to reveal the mechanisms behind the disease as well as develop therapeutics. The siRNA research is part of a three-part strategy which also includes repurposing drugs for treating FLC and using molecules known as PROTACs to selectively degrade the DNAJB1::PRKACA protein that drives FLC. The drug repurposing is in advanced preparation for a clinical trial, and some of the PROTAC work is part of an international collaboration that was just awarded a $25 million Cancer Grand Challenges grant to develop treatments for high-risk, oncogene-driven cancers in children.
Recently, Simon’s lab discovered that they could completely stop FLC tumor growth in mice using shRNAs, short sequences of RNA that can be engineered to disrupt mRNA. The approach also caused many tumors to shrink or disappear.
Having shown that quelling the fused oncogene killed tumor cells, they decided to experiment with derailing kinase A activity, which research indicated was driving tumor growth.
Unlocking the cell door
The problem was, fused kinase A and its “wild type” form are nearly identical, so “any drug you developed that blocks fused kinase A activity would affect all kinase A, including in normal cells,” Neumayer says. “In other words, you would incur a host of problematic side effects.”
They had to find a way to disable toxic kinase while containing the impact so that kinase in other cells weren’t affected. To do that, they specifically designed siRNAs to aim at the fusion. But because siRNAs cannot cross cell membranes, the researchers needed to figure out a way to get the siRNA inside.
Luckily, a door into the cell had already been identified: a receptor called ASGR1 that’s only expressed in liver cells. Its key had been too: a binding molecule known as GalNAc. Together, the duo function as a waste disposal team.
GalNAc conjugate therapies, which attach siRNA molecules to the ligand to deliver therapies inside cells, are already on the market for conditions such as hereditary transthyretin-mediated amyloidosis and atherosclerotic cardiovascular disease. The team wondered if they could use the same approach to thwart the kinase driving FLC tumors.
Stabilization and shrinkage
To test their theory, the researchers attached a custom siRNA to the receptor’s ligand and studied the method in a variety of tumor models in mice and in human cell cultures. The conjugate therapy was not only successfully delivered into the cells in all contexts, but it also caused a reduction of mRNA in the oncogene, preventing the production of the protein and resulting in the tumors stagnating or shrinking, unable to grow. They also did not detect any liver toxicity in the mice, indicating the animals tolerated the siRNAs well.
“What we saw was tumor inhibition, when we had hoped to outright kill the tumor,” Neumayer notes. “Our future direction will be to try to figure out how to improve that.”
They also tested the specificity of the siRNA by injecting it into tumor cells of another type of liver cancer. It had no toxic impact on them—exactly as the researchers hoped. “We wanted to show that our siRNA is so targeted to the FLC fusion oncogene that it has no side effects on other cells—even on other cancer cells,” he notes.
Neumayer says the findings suggest that siRNAs may be effective beyond FLC, capable of treating tumors throughout the body with a high degree of specificity: “I think siRNA medicines as a class will have a big impact over time as a new kind of genetic therapy.”