Technology Pipeline and Policies
From 2015 to 2017, the G10K consortium worked with the major sequencing and assembly companies (e.g., Illumina, Pacific Biosciences, Oxford Nanopore, Bionano, 10X Genomics, NRGene, Dovetail Genomics, Phase Genomics, Arima Genomics), major sequencing centers (BGI, Broad Institute, Sanger Institute, Washington University Genome Center), major public genome archive and annotation centers (NCBI, Ensembl, UCSC), and experts in academia and government (NIH, NSF) to test, improve, and generate new approaches for producing the highest quality, error-free, 3rd generation reference genome assemblies achievable for the least cost possible. For the first time, we tested each of these technologies on one individual animal, a bird (hummingbird or zebra finch) and a mammal (goat or human), such that our analyses were not hampered by the common problem of multiple variables changing simultaneously, which has plagued previous comparative genome technology efforts.
Our goal with these approaches is to generate a genome with a metric minimum contig N50 of 1 million bp (1Mb), scaffold N50 of 10Mb, 90% of the genome assembled into chromosomes confirmed by 2 independent sources, a base-call quality error of QV40 (no more than 1 nucleotide error in 10,000 bp), and haplotype phased. We call this a 3.4.2.QV40 phased metric, where the first three numbers are the exponents of the N50 contig, N50 scaffold, and level of chromosomal assembly.
A major challenge for saving species and conducting high-quality genomic research has been finding cost-effective and sufficient technology to generate high-quality genomes. We worked with industry partners to develop unprecedented high-resolution genome sequencing methods at significantly lower costs than current, less robust technologies.
The Current Pipeline
The current pipeline (Figure 1) to meet the 3.4.2.QV40 phased metric with the fewest errors currently achievable consists of a combination of the following approaches:
- 60X PacBio long-reads for an initial phased contig assembly (30X/haplotype);
- 68X coverage of 10X Genomics-linked reads for intermediate-range scaffolding and further phasing (34X/haplotype);
- 80X Bionano optical maps to correct scaffolding errors and for further scaffolding;
- 68X Hi-C linked reads for long-range scaffolding;
- PacBio Jelly algorithm to fill gaps using long-reads;
- 10X Genomics Illumina short-reads for base-call accuracy polishing; and
- Assembly algorithms that merge multiple data types without creating haplotype errors
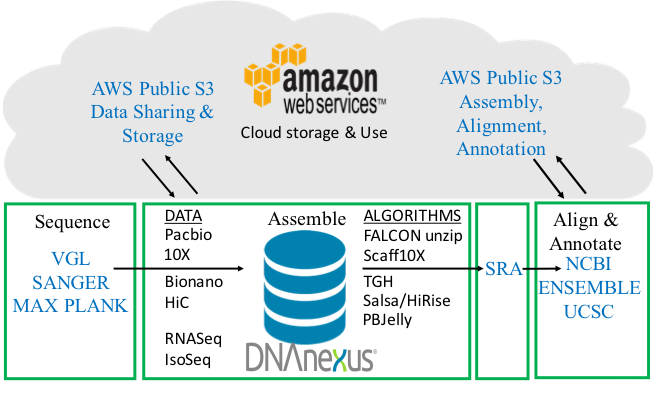
Full VGP pipeline. From sample processing, sequence data generation, assembly algorithms, annotation, to uploading onto public databases. The pipeline is a 4-way partnership between the sequence data generators and assemblers, DNAnexus, AWS, and annotation centers. Figure modified from Mark Mooney (DNAnexus).
Rationale
High-quality error-free genome assemblies and annotations are necessary as current 1st and 2nd generation genome sequencing approaches generate numerous errors that cause a variety of problems in downstream analyses. Parts of genes are missing, and some are incorrectly assembled, while others are completely missing from the assemblies despite pieces found in the raw sequence reads. Due to these fragmented, error-prone assemblies, researchers have had to clone, re-sequence, and correct individual genes. In some cases, the gene structures are too complex, too long, or too closely related, preventing even the Sanger-based higher quality 1st generation methods from correcting genome assemblies. In many other instances, investigators do not even know that they are working with incorrect gene sequences and structures, impacting many scientific findings and scientific progress.