Faster-acting antidepressants may finally be within reach
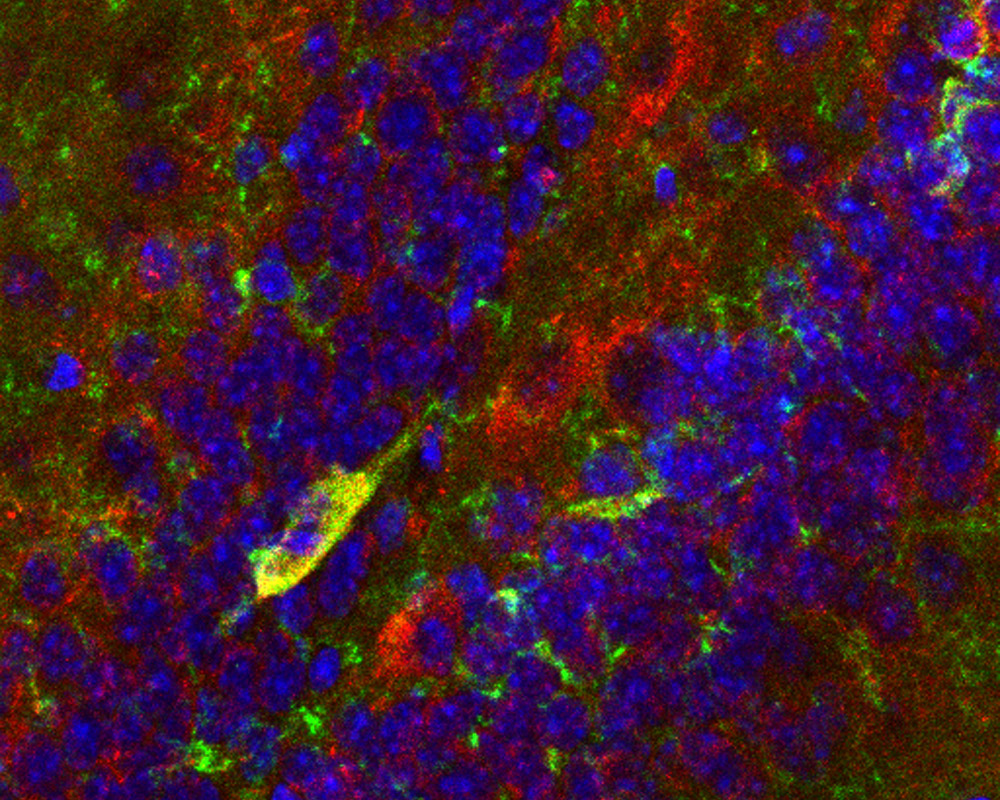
CCK neurons (red) within the mouse dentate gyrus. The cells produce p11 (green), a protein the researchers showed is necessary for SSRI action.
Some activity patterns in the brain can be dangerous, producing persistent dark moods that drain people’s motivation, pleasure, and hope. For the past thirty years, pills like Prozac or Zoloft—collectively known as selective serotonin reuptake inhibitors, or SSRIs—have offered millions of Americans a way to shed the heavy cloak of depression and attain more wholesome states of mind.
These medications were designed to increase nerve cells’ access to serotonin, a chemical that helps the brain regulate certain emotions. Yet researchers still don’t know precisely how the drugs work to adjust errant brain chemistry, or how to make them work better.
Now, a team of Rockefeller scientists has for the first time described how SSRIs initiate their action by targeting a particular type of nerve cell. Their findings, published last week in Neuron, may provide a path to new antidepressants that would not only be safer to use than existing ones, but that would also act more quickly.
Lucian Medrihan, a research associate in the lab of neuroscientist and Nobel laureate Paul Greengard who led the study, explains that while existing SSRIs can produce moderate effects within hours or even minutes, most people don’t really begin to feel better until they’ve been on the drugs for a significant amount of time—a major drawback when it comes to treating clinical depression. The drugs may also cause a wide range of uncomfortable side effects, including nausea, dizziness, weight gain, and sexual dysfunction.
Finding the cells that matter
The basic idea behind SSRIs is relatively simple. When a neuron releases serotonin to signal another cell, it normally reabsorbs excess amounts of the neurotransmitter, preventing it from lingering in the space where the two nerve cells meet. The drugs interfere with this mopping-up step, essentially prolonging the signal.
What happens next has been a hard nut for neuroscientists to crack, however, because of the intrinsic complexity of the brain.
At least 1000 types of neurons could potentially be affected by a surge in serotonin, and they don’t all respond in the same way—some get triggered, for example, while others calm down. “That’s because there are 14 types of serotonin receptors present in various combinations in different neurons,” says Yotam Sagi, a senior research associate in Greengard’s lab. How a cell reacts to the neurotransmitter depends on the particular hodgepodge of receptors it carries.
Sagi and Medrihan set out to identify the earliest molecular steps by which SSRIs curb depression. To narrow their search, they honed in on a region of the brain known as the dentate gyrus, and on a particular group of cells called cholecystokinin (CCK)-expressing neurons, which they suspected were affected by SSRI-induced serotonin changes.
Using a technique called translating ribosome affinity purification, developed at Rockefeller by Nathaniel Heintz and Greengard, Sagi was able to identify the serotonin receptors present on CCK cells. “We were able to show that one type of receptor, called 5-HT2A, is important for SSRIs’ long-term effect,” he says, “while the other, 5-HT1B, mediates the initiation of their effect.”
A potential first step to relieving depression
Next, Medrihan set up a series of intricate experiments to see if he could mimic an SSRI response by manipulating CCK neurons in living mice. He suppressed the activity of these cells with chemogenetics, a technique that makes it possible to switch nerve cells on or off at will, and placed panels of tiny electrodes inside mouse brains. He then monitored the firing of other neurons in the dentate gyrus. “Only five years ago, this research would not have been feasible,” he says of the methods involved.
The results were unmistakable: when a mouse’s CCK neurons were inhibited, the same neural pathways that mediate responses to SSRIs lit up. In targeting these cells, the scientists had seemingly recreated a quickened, Prozac-like response without the drug itself.
They also performed behavioral experiments by placing the mice in a pool and monitoring their swimming patterns. After the CCK neurons had been briefly silenced, the behavior of these animals, which had not received any drugs, was similar to that seen in other mice after Prozac treatment: they swam with prolonged zest.
Greengard, who is Vincent Astor Professor, says the research resolves an important question in the field. “Many different types of synapses throughout the brain use serotonin as their neurotransmitter,” he says. “An issue of major importance has been to identify where in the myriad of neurons the antidepressants initiate their pharmacological action.”
The findings, which identify CCK neurons in the dentate gyrus as the site of interest, will advance scientists’ understanding of how SSRI antidepressants work, and “should also facilitate development of new classes of potent and selective drugs,” Greengard says. Such future therapies would presumably act faster than existing SSRIs, and might also produce fewer side effects.



